| The HAPLOTYPE Procedure |
| PROC HAPLOTYPE Statement |
- PROC HAPLOTYPE <options> ;
You can specify the following options in the PROC HAPLOTYPE statement.
- ALPHA=number
specifies that a confidence level of 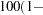 number
number  is to be used in forming the confidence intervals for estimates of haplotype frequencies. The value of number must be between 0 and 1, inclusive, and 0.05 is used as the default value if it is not specified.
is to be used in forming the confidence intervals for estimates of haplotype frequencies. The value of number must be between 0 and 1, inclusive, and 0.05 is used as the default value if it is not specified.
-
BURNIN=number
Experimental indicates that number iterations are discarded as burn-in when EST=BAYESIAN is specified. The value of number cannot be greater than the value specified in the TOTALRUN= option and must be greater than 0. The default is
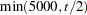 , where
, where  is the number of total runs.
is the number of total runs. - CONV=number
specifies the convergence criterion for iterations of the EM algorithm, where
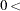 number
number 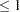 . The iteration process is stopped when the ratio of the change in the log likelihoods to the former log likelihood is less than or equal to number for the number of consecutive iterations specified in the NLAG= option (or 1 by default), or after the number of iterations specified in the MAXITER= option has been performed. The default value is 0.00001.
. The iteration process is stopped when the ratio of the change in the log likelihoods to the former log likelihood is less than or equal to number for the number of consecutive iterations specified in the NLAG= option (or 1 by default), or after the number of iterations specified in the MAXITER= option has been performed. The default value is 0.00001. - CUTOFF= number
specifies a lower bound on a haplotype’s estimated frequency in order for that haplotype to be included in the "Haplotype Frequencies" table. The value of number must be between 0 and 1, inclusive. By default, all possible haplotypes from the sample are included in the table.
- DATA=SAS-data-set
names the input SAS data set to be used by PROC HAPLOTYPE. The default is to use the most recently created data set.
- DELIMITER=’string’
indicates the string that is used to separate the two alleles that compose the genotypes contained in the variables specified in the VAR statement. This option is ignored if GENOCOL is not specified.
- EST=BAYESIAN
- EST=EM
- EST=STEPEM
indicates the method to be used for estimating haplotype frequencies. By default or when EST=EM is specified, the EM algorithm is used. When EST=STEPEM, the stepwise EM algorithm is used to calculate estimates of haplotype frequencies. When EST=BAYESIAN, a Bayesian method is used for estimating haplotype frequencies.
- GENOCOL
indicates that columns specified in the VAR statement contain genotypes instead of alleles. When this option is specified, there is one column per marker. The genotypes must consist of the two alleles separated by a delimiter. For a genotype with one missing allele, use a blank space to indicate a missing value; if both alleles are missing, either use a single missing value for the entire genotype or use the delimiter alone.
- INDIVIDUAL=variable
- INDIV=variable
specifies the individual ID variable when using the TALL option. This variable can be character or numeric.
- INIT=LINKEQ
- INIT=RANDOM
- INIT=UNIFORM
indicates the method of initializing haplotype frequencies to be used in the EM algorithm. INIT=LINKEQ initializes haplotype frequencies assuming linkage equilibrium by calculating the product of the frequencies of the alleles that compose the haplotype. INIT=RANDOM initializes haplotype frequencies with random values from a Uniform(0,1) distribution, and INIT=UNIFORM assigns equal frequency to all haplotypes. By default, INIT=LINKEQ.
-
INTERVAL=number
Experimental indicates that the non-burn-in iterations of the Bayesian estimation method when EST=BAYESIAN is specified are thinned by only recording the result from every number iterations. The value of number must be greater than 0, and the default is 1 (every iteration is used).
- ITPRINT
requests that the "Iteration History" table be displayed. This option is ignored if the NOPRINT option is specified.
- LD
requests that haplotype frequencies be calculated under the assumption of no LD, in addition to being calculated using the EM algorithm. When this option is specified, the "Test for Allelic Associations" table is displayed, which contains statistics for the likelihood ratio test for allelic associations. This option is ignored if the NOPRINT option is specified.
- MARKER=variable
specifies the marker ID variable when using the TALL option. This variable contains the names of the markers that are used in all output and can be character or numeric.
- MAXITER=number
specifies the maximum number of iterations to be used in the EM algorithm. The number must be a nonnegative integer. Iterations are carried out until convergence is reached according to the convergence criterion or until number iterations have been performed. The default is MAXITER=100.
- NDATA=SAS-data-set
names the input SAS data set containing names, or identifiers, for the markers used in the output. There must be a NAME variable in this data set, which should contain the same number of rows as there are markers in the input data set specified in the DATA= option. When there are fewer rows than there are markers, markers without a name are named using the PREFIX= option. Likewise, if there is no NDATA= data set specified, the PREFIX= option is used. Note that this data set is ignored if the TALL option is specified in the PROC HAPLOTYPE statement. In that case, the marker variable names are taken from the marker ID variable specified in the MARKER= option.
- NLAG=number
specifies the number of consecutive iterations that must meet the convergence criterion specified in the CONV= option (0.00001 by default) for the iteration process of the EM algorithm to stop. The number must be a positive integer. If this option is omitted, one iteration must satisfy the convergence criterion by default.
- NOPRINT
suppresses the display of the "Analysis Information," "Iteration History," "Haplotype Frequencies," and "Test for Allelic Associations" tables. Either the OUT= option, the TRAIT statement, or both must be used with the NOPRINT option.
- NSTART=number
specifies the number of different starts used for the EM algorithm. When this option is specified, PROC HAPLOTYPE starts the iterations with different random initial values number
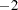 times as well as once with uniform frequencies for all the haplotypes and once using haplotype frequencies assuming linkage equilibrium (independence). Results on the analysis, using the initial values that produce the best log likelihood, are then reported. The number must be a positive integer. If this option is omitted or NSTART=1, only one start with initial frequencies generated according to the INIT= option is used.
times as well as once with uniform frequencies for all the haplotypes and once using haplotype frequencies assuming linkage equilibrium (independence). Results on the analysis, using the initial values that produce the best log likelihood, are then reported. The number must be a positive integer. If this option is omitted or NSTART=1, only one start with initial frequencies generated according to the INIT= option is used. - OUT=SAS-data-set
names the output SAS data set containing the probabilities of each genotype being resolved into all of the possible haplotype pairs.
- OUTCUT=number
specifies a lower bound on a haplotype pair’s estimated probability given the individual’s genotype in order for that haplotype pair to be included in the OUT= data set. The value of number must be between 0 and 1, inclusive. By default, number
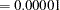 . In order to be able to view all possible haplotype pairs for an individual’s genotype, OUTCUT=0 can be specified.
. In order to be able to view all possible haplotype pairs for an individual’s genotype, OUTCUT=0 can be specified. - OUTID
indicates that the variable _ID_ created by PROC HAPLOTYPE should be included in the OUT= data set in addition to the variable(s) listed in the ID statement. When the ID statement is omitted, this variable is automatically included. This option is ignored when the TALL option is used.
- PREFIX=prefix
specifies a prefix to use in constructing names for marker variables in all output. For example, if PREFIX=VAR, the names of the variables are VAR1, VAR2, ..., VARn. Note that this option is ignored when the NDATA= option is specified, unless there are fewer names in the NDATA data set than there are markers; it is also ignored if the TALL option is specified, in which case the marker variable names are taken from the marker ID variable specified in the MARKER= option. Otherwise, if this option is omitted, PREFIX=M is the default when variables contain alleles; if GENOCOL is specified, then the names of the variables specified in the VAR statement are used as the marker names.
- SE=BINOMIAL
- SE=JACKKNIFE
specifies the standard error estimation method when the EM or stepwise EM algorithm is used for estimating haplotype frequencies. There are two methods available: the BINOMIAL option, which gives a standard error estimator from a binomial distribution and is the default method, and the JACKKNIFE option, which requests that the jackknife procedure be used to estimate the standard error.
- SEED=number
specifies the initial seed for the random number generator used for creating the initial haplotype frequencies when INIT=RANDOM and/or to permute the data when the PERMUTATION= option of the TRAIT statement is specified. The value for number must be an integer; the computer clock time is used if the option is omitted or an integer less than or equal to 0 is specified. For more details about seed values, see SAS Language Reference: Concepts.
- STEPTRIM=number
indicates the cutoff to be used for the stepwise EM algorithm when trimming the haplotype table, where
 . This option is implemented only when EST=STEPEM is specified. By default, this number is set to
. This option is implemented only when EST=STEPEM is specified. By default, this number is set to 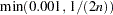 , where
, where  is the number of individuals in the data set.
is the number of individuals in the data set. - TALL
indicates that the input data set is of an alternative format. This format contains the following columns: two containing marker alleles (or one containing marker genotypes if GENOCOL is specified), one for the marker identifier, and one for the individual identifier. The MARKER= and INDIV= options must also be specified in order for this option to be in effect. Note that when this option is used, the DATA= data set must first be sorted by any BY variables, then sorted by the marker ID variable, and then sorted by the individual ID variable.
-
THETA=number
Experimental requests that number be used as the scaled mutation rate
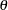 when EST=BAYESIAN instead of the default, which is
when EST=BAYESIAN instead of the default, which is 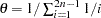 for a sample of individuals. This value must be positive.
for a sample of individuals. This value must be positive. -
TOTALRUN=number
Experimental - TOT=number
indicates the total number of iterations to use when EST=BAYESIAN, including the burn-in. The value of number must be greater than 0, and the default is 10,000.
Copyright © 2008 by SAS Institute Inc., Cary, NC, USA. All rights reserved.