| The HAPLOTYPE Procedure |
| Missing Values |
An individual’s 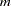 -locus genotype is considered to be partially missing if any, but not all, of the alleles are missing. Genotypes with all missing alleles are dropped from calculations for haplotype frequencies, although these individuals can still be used as described in the following paragraph. Also, if there are any markers with all missing values in a BY group (or the entire data set if there is no BY statement), no calculations are performed for that BY group. Partially missing genotypes are used in the EM algorithm and the jackknife procedure. In calculating the allele frequencies, missing alleles are dropped and the frequency of an allele
-locus genotype is considered to be partially missing if any, but not all, of the alleles are missing. Genotypes with all missing alleles are dropped from calculations for haplotype frequencies, although these individuals can still be used as described in the following paragraph. Also, if there are any markers with all missing values in a BY group (or the entire data set if there is no BY statement), no calculations are performed for that BY group. Partially missing genotypes are used in the EM algorithm and the jackknife procedure. In calculating the allele frequencies, missing alleles are dropped and the frequency of an allele  at a marker is obtained as the number of alleles in the data divided by the total number of nonmissing alleles at the marker in the data. In the E step of the EM algorithm, the frequency of a partially missing genotype is updated for every possible genotype. In the M step, haplotypes resulting from a missing genotype can bear some missing alleles. Such a haplotype is not considered as a new haplotype, but rather all existing haplotypes that have alleles identical to the nonmissing alleles of this haplotype are updated. Dealing with missing genotypes involves looping through all possible genotypes in the E step and all possible haplotypes in the M step. The stepwise EM algorithm performs a series of two-step processes involving EM estimation followed by trimming the set of haplotypes. Thus, in the EM estimation step, missing values are handled as described for the EM algorithm. Depending on the input data set, missing genotypes can increase the computation time substantially for either estimation method.
at a marker is obtained as the number of alleles in the data divided by the total number of nonmissing alleles at the marker in the data. In the E step of the EM algorithm, the frequency of a partially missing genotype is updated for every possible genotype. In the M step, haplotypes resulting from a missing genotype can bear some missing alleles. Such a haplotype is not considered as a new haplotype, but rather all existing haplotypes that have alleles identical to the nonmissing alleles of this haplotype are updated. Dealing with missing genotypes involves looping through all possible genotypes in the E step and all possible haplotypes in the M step. The stepwise EM algorithm performs a series of two-step processes involving EM estimation followed by trimming the set of haplotypes. Thus, in the EM estimation step, missing values are handled as described for the EM algorithm. Depending on the input data set, missing genotypes can increase the computation time substantially for either estimation method.
When the TRAIT statement is specified, any observation with a missing trait value is dropped from calculations used in the tests for marker-trait association and haplotype-trait associations. However, observations with missing trait values are included in calculating the frequencies shown in the "Haplotype Frequencies" table, which are then used in the OUT= data set. The combined frequencies listed in the "Tests for Haplotype-Trait Association" table might therefore be different from these frequencies in this situation. Also, if an individual is missing all alleles but has a nonmissing trait value, the individual is included in the permutations of the trait value when PERMS= is specified in the TRAIT statement.
Copyright © 2008 by SAS Institute Inc., Cary, NC, USA. All rights reserved.